

Peptide Fundamentals: Key Takeaways
Peptides bridge the gap between small molecules and large biologics. They can target specific receptors with high selectivity. And the cost of manufacturing them is lower than that of monoclonal antibodies.
Data shows that more than 120 therapeutic peptide and protein-based drugs had been approved globally by 2023. The combined peptide and biologic drug sales exceed $500 billion annually.
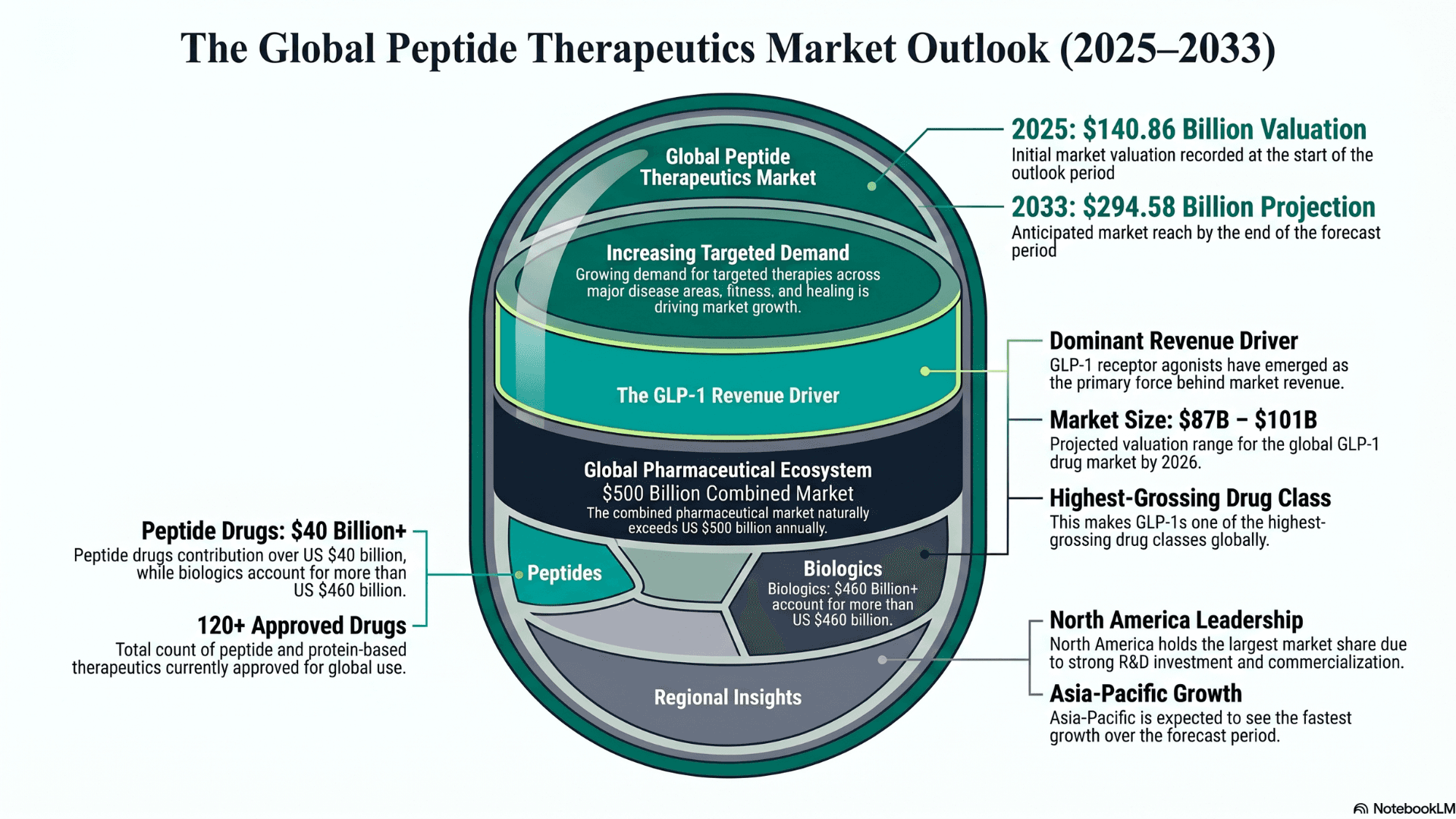
The peptide market was valued at approximately US$ 140.86 billion in 2025 and is projected to reach around US$ 294 billion by 2033. The demand for GLP-1 receptor agonists is pushing this growth.
The two main issues with peptide drug development are rapid enzymatic degradation and poor membrane permeability. This pushes developers towards chemical modification strategies, including cyclization, D-amino acid substitution, PEGylation, lipidation, and Fc-fusion.
Finally, as of 2026, there are no AI-generated peptides that have been approved for commercial use. But there is significant development in the tech.
From Biology to Therapeutics: How a Century of Peptide Science Became a Global Industry
What was the first therapeutic peptide, and how did the field develop from there?
Peptides have a fascinating history. It begins with the discovery of insulin. Frederick Banting and colleagues first extracted insulin from animal pancreatic tissue in 1921 and validated it in a 12-year-old patient in 1922 (Xiao et al., Nature).
Then, in 1923, Eli Lilly and Company released the first commercial insulin product. Due to the tech constraints, limited research, and other factors, insulin was derived from cows and pigs for 90 years. Unfortunately, this also triggered various immune responses in some patients. The non-human protein sequences were activating antibody production.
Peptide development only began to gain steam in the 1950s. Key breakthroughs in this period included Vincent du Vigneaud’s synthesis of oxytocin and vasopressin. Then Robert Merrifield developed solid-phase peptide synthesis. These not only allowed large-scale production but also led to early peptide drug approvals.
A significant development was recombinant human insulin. Recombinant means inserting the human insulin gene into a host cell so it produces the protein using genetic engineering. Approved by the FDA in 1982 as the first recombinant therapeutic protein, it directly addressed the problem of animal-derived insulin. Goserelin was approved for cancer in 1989. Enfuvirtide was approved for HIV in 2003.
The twenty-first century has seen a rapid growth in this field. The use of novel design techniques, display library technology, and advanced delivery systems transformed peptide drug development. Over the past two decades, the number of approved peptide drugs has surpassed 60 globally, with more approval processes underway (Xiao et al., Nature).
Among the most significant milestones was the 2019 approval of oral semaglutide, the first GLP-1 receptor agonist approved for oral administration. The significance of this cannot be understated. Global peptide research has only grown from that point onwards.
How large is the peptide drug market, and where is it heading?
Now that is a conversation that can’t wait. If we look at market data, the global peptide therapeutics market was valued at approximately USD 140.86 billion in 2025. Now it is projected to reach USD 294.58 billion by 2033, growing at a CAGR of 8.73% (Grand View Research).
There is an increasing demand for targeted therapies using peptides across major disease areas, fitness, healing, etc. This is driving the growth. It is no surprise that GLP-1 receptor agonists have emerged as the dominant revenue driver. The global GLP-1 drug market is estimated to be between $87 billion and $101 billion in 2026. This makes it one of the highest-grossing drug classes globally.
North America continues to hold the largest market share due to strong R&D investment and commercialization. But Asia-Pacific is expected to see the fastest growth over the forecast period.
Just so we have the entire picture, more than 120 peptide and protein-based drugs have been approved globally. Peptide drugs are contributing more than US$ 40 billion, and biologics more than US$ 460 billion. The combined pharmaceutical market is naturally exceeding US$ 500 billion annually. To understand why this growth is happening, we need to look at how peptides compare to other drug types.
Why do peptide drugs hold an advantage over both small molecules and large biologics?
There are three main types of drugs in medicine today. Each has its own strengths and weaknesses. Seeing where peptides fit in shows why drug companies are investing so much money in them.
Small molecule drugs:
They are cheap to produce and can be swallowed as pills. But their size is their limiting factor. Protein-protein interactions in the body involve contact surfaces covering 1,500 to 3,000 square angstroms.
But a small molecule drug only covers 300 to 1,000 square angstroms of protein surface. This does mean that it is unable to block large protein-to-protein interactions effectively (Wang et al., Nature).
Large biologic drugs:
These are primarily monoclonal antibodies that are large enough to cover these interaction surfaces. In this case, size is not an advantage. They are more than 150 kilodaltons in molecular weight. This means that their tissue penetration is limited, and that pushes the manufacturing cost upwards.
Peptides:
Finally, we have the solution. Peptides are big enough to stop complex protein interactions. But more importantly, they are small enough for deeper tissue penetration. Thus, they have lower manufacturing costs than monoclonal antibodies.
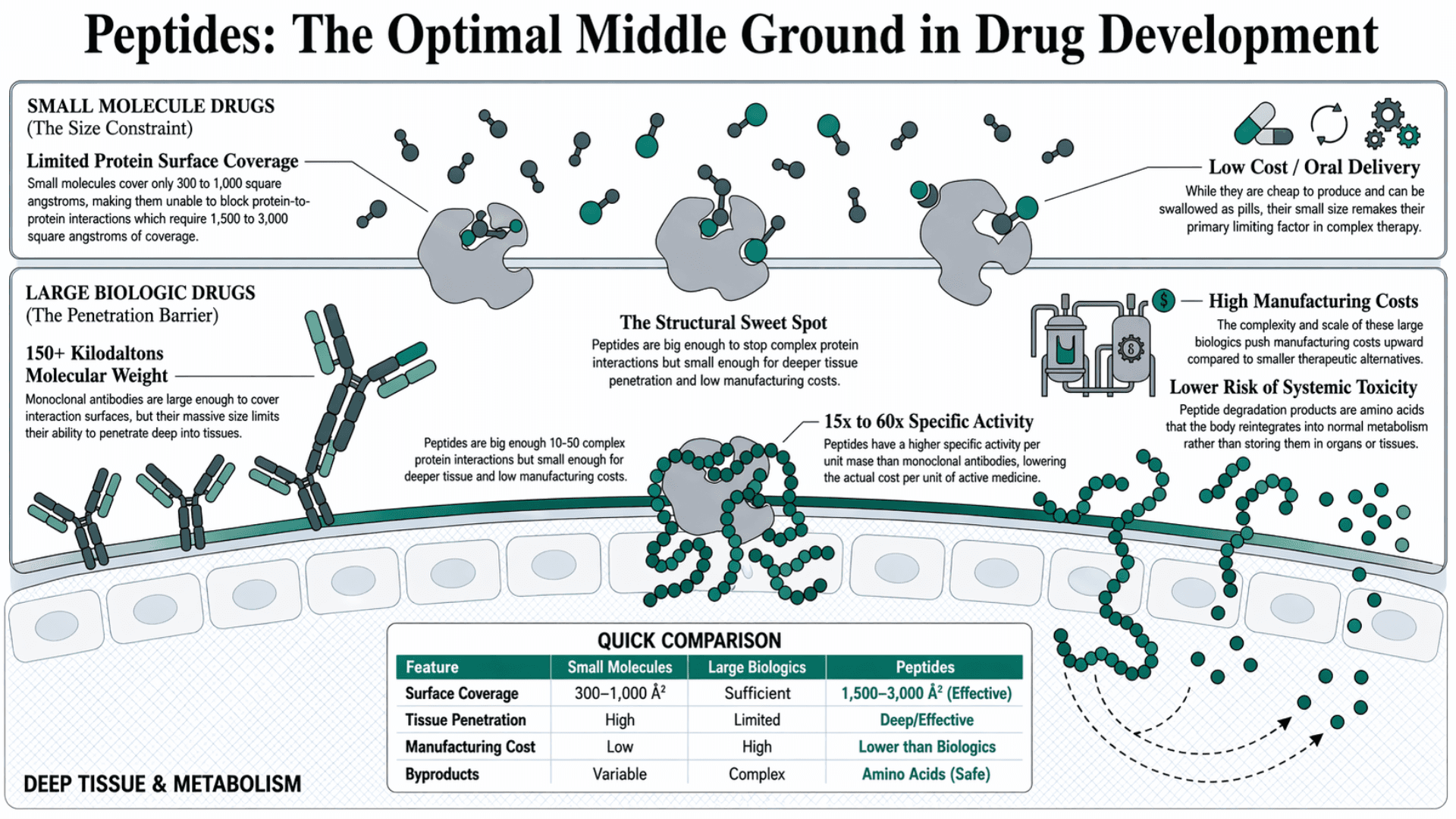
Therapeutic peptides typically have a length of 10 to 50 amino acids. Their higher specific activity per unit mass, estimated at 15 to 60 times that of monoclonal antibodies, lowers the actual cost per unit of active medicine (Xiao et al., Nature).
A 2022 comprehensive review published in Nature's Signal Transduction and Targeted Therapy confirmed that peptides also carry a lower risk of systemic toxicity. Their degradation products are amino acids, which the body reintegrates into normal cellular metabolism (Wang et al., Nature). This means peptide degradation byproducts do not get stored in organs or tissues.
Mechanism of Action: How Therapeutic Peptides Engage Their Targets
Therapeutic peptides work by acting as chemical keys. They bind directly to specific receptors on the surface of cells and alter the signaling pathways connected to those receptors. Agonistic peptides activate and enhance downstream cellular functions. Antagonistic peptides block or suppress those functions (Fetse et al., NIH).
It has also been confirmed that peptides can also access intracellular targets through clathrin-mediated endocytosis or clathrin-independent endocytosis. This phenomenon occurs when their extracellular targets alone are not sufficient.
Clathrin-mediated endocytosis is when a cell pulls something inside by wrapping it in a protein coat called clathrin. And clathrin-independent endocytosis is when the cell takes something in without using that clathrin coat, through other entry pathways.
For extracellular targets, the peptide attaches to a specific site on the receptor. This causes the receptor to change its shape, which then sends a signal inside the cell and triggers a response.
More than 90% of peptides currently in active clinical development target extracellular receptors, including G protein-coupled receptors and the GLP-1 receptor (Wang et al., Nature).
The reason for extracellular targets being the focus exposes the reality of the situation. To reach intracellular targets, peptides need to cross cell membranes. That is a considerable challenge for most peptide sequences.
How does the physical structure of a peptide determine its receptor binding?
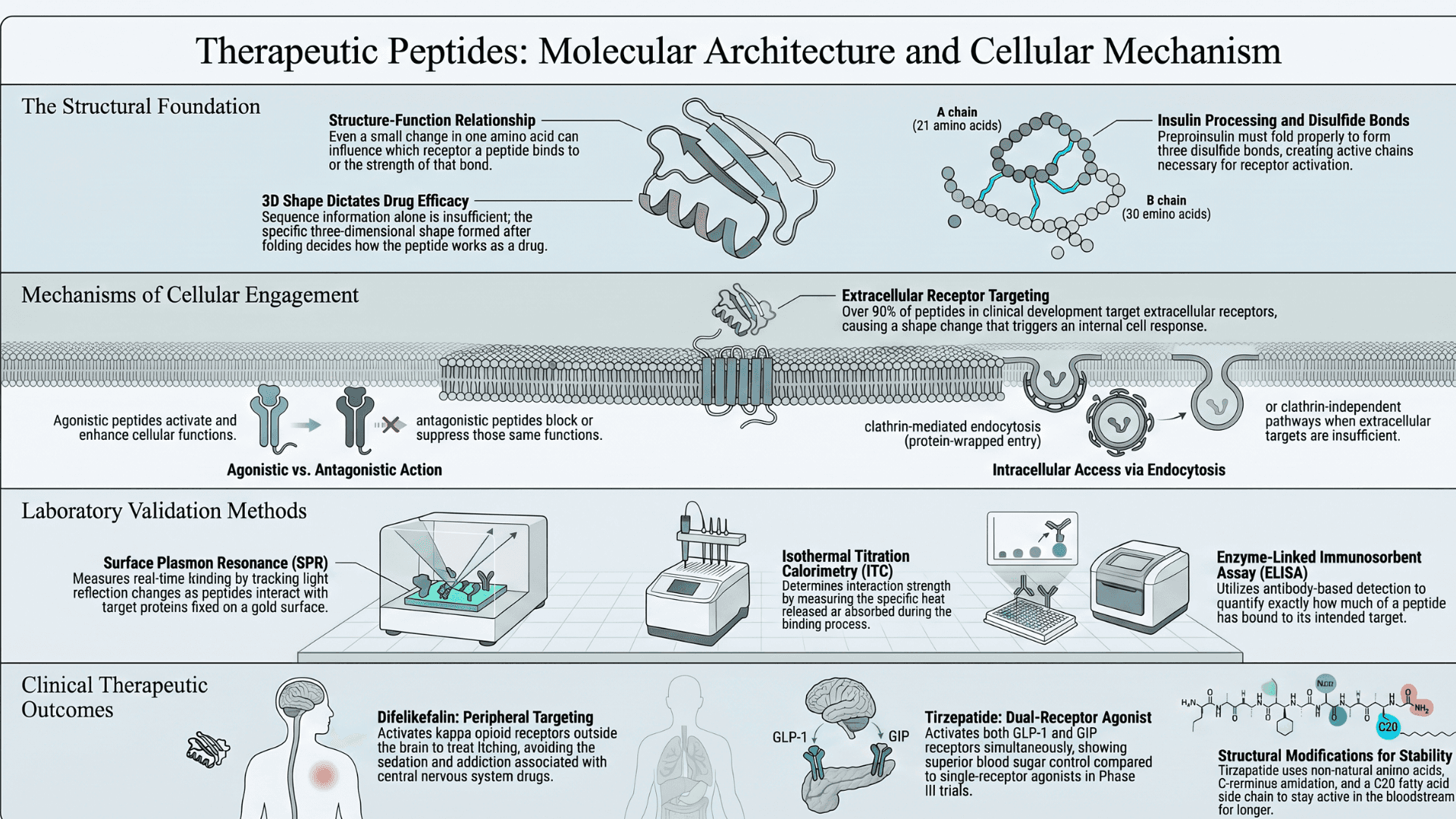
Peptide structures and biological functions are directly related to each other. Even a small change in one amino acid can influence which receptor the peptide will choose to bind to. Or how strong the bond will be. This can be clearly seen in the processing of insulin.
Insulin is first made as an inactive form called preproinsulin. The signal peptide of this molecule is removed. This allows it to fold properly and form three disulfide bonds. It is then further processed into its active form, which has two chains. The A chain has 21 amino acids, and the B chain has 30. These two chains are then connected by disulfide bonds (strong links between two sulfur atoms in a protein that help hold its shape together).
This structure is necessary for insulin to bind to its receptor on the cell surface. Once it binds, the receptor activates itself. This results in sending a signal inside the cell, which then helps control the blood sugar levels. It is important to note that the disulfide bonds need to form correctly. Otherwise, the insulin won’t be able to maintain its shape, thus failing to connect to the receptor (Iglesias et al., NIH).
This proves that for developing a drug, sequence information isn’t enough. The three-dimensional shape of the peptide, formed after it folds and is processed in the body, decides how it works as a drug.
What laboratory methods are used to measure receptor binding strength?
There is a reason why so few peptide drugs have been approved. Even before it enters clinical testing, scientists need to look at two things. How strongly and how specifically it binds to its target. The 2025 regulatory guidelines show three main techniques used to do this (Elsayed et al., NIH).
Surface Plasmon Resonance (SPR):
This measures binding in real time by tracking changes in light reflection when a peptide interacts with a target protein fixed on a gold surface.
As binding occurs, the change in mass affects the reflected light signal. This produces a curve that shows how quickly the peptide attaches and detaches. It also shows how strong the interaction is.
Isothermal Titration Calorimetry (ITC):
It measures the heat released or absorbed during binding. This heat signal shows how strong the interaction is.
Enzyme-Linked Immunosorbent Assay (ELISA):
This uses antibody-based detection to check how much of the peptide has bound to its target. Each of these methods serves a clear purpose in providing information. Together, they showcase how peptide bonds behave.
What specific clinical examples show how receptor binding translates to therapeutic outcomes?
To understand how unique clinical outcomes can be when there is precise receptor targeting, we can look at two peptides approved by the FDA.
Difelikefalin was approved in 2021. It is used to treat severe itching in people with chronic kidney disease. It works by activating a specific type of receptor called the kappa opioid receptor. These receptors are located outside the brain, in the body (peripheral receptors).
The drug mainly acts outside the brain and not in the central nervous system. This means side effects like sedation and addiction, which are common with opioid drugs, are largely absent (Fetse et al., NIH).
Tirzepatide was approved in 2022 for type 2 diabetes. It activates both the GLP-1 receptor and the GIP receptor at the same time. This means both working together to improve blood sugar control. In the SURPASS Phase III clinical trials, tirzepatide demonstrated superior performance over single-receptor agonists, including dulaglutide and semaglutide (Xiao et al., Nature).
It is made of a chain of 39 building blocks (amino acids). Two of these building blocks are slightly modified (non-natural). They are placed at positions 2 and 13 to make the drug more stable in the body. The end of the molecule is chemically adjusted (amidated at the C-terminus) to help it stay active for longer.
A long fatty chain (C20 side chain) is attached at position 20 using a linker. This helps the drug stay in the bloodstream for a longer time. All of these changes make the drug last longer in the body and bind more effectively to its target receptors.
How do positively charged peptides enter cells when intracellular targets must be reached?
As discussed earlier, peptides face the cell membrane barrier every time they want to reach the intracellular targets. And they have two entry points.
Clathrin-mediated endocytosis:
This is the more common way peptides enter a cell. Certain areas of the cell membrane form deep pockets around the peptide. These pockets then close off and form small bubbles (vesicles) that carry the peptide inside the cell. This process uses energy from the cell itself.
Clathrin-independent endocytosis:
This is another way peptides enter the cell. But it does not use the clathrin protein. Instead, the cell uses different parts of its membrane, such as lipid-rich areas and small curved structures called caveolae. These also pinch off from the surface and carry the peptide inside the cell (Fetse et al., NIH), (Xiao et al., Nature).
But we would be remiss not to mention another mechanism adopted by cell-penetrating peptides. It’s called direct translocation. This path doesn’t require energy. The peptide directly interacts with the lipid layer of the membrane. It creates small openings, loosens the membrane structure, or creates short-lived transport mechanisms that help it move inside.
The same peptide can switch between endocytosis and direct translocation depending on its concentration, the type of cargo it carries, and the specific cell type it encounters (Wang et al., Nature).
Design Complexity: Engineering a Peptide That Survives the Human Body
What synthesis method is used to build therapeutic peptides, and what are its limits?
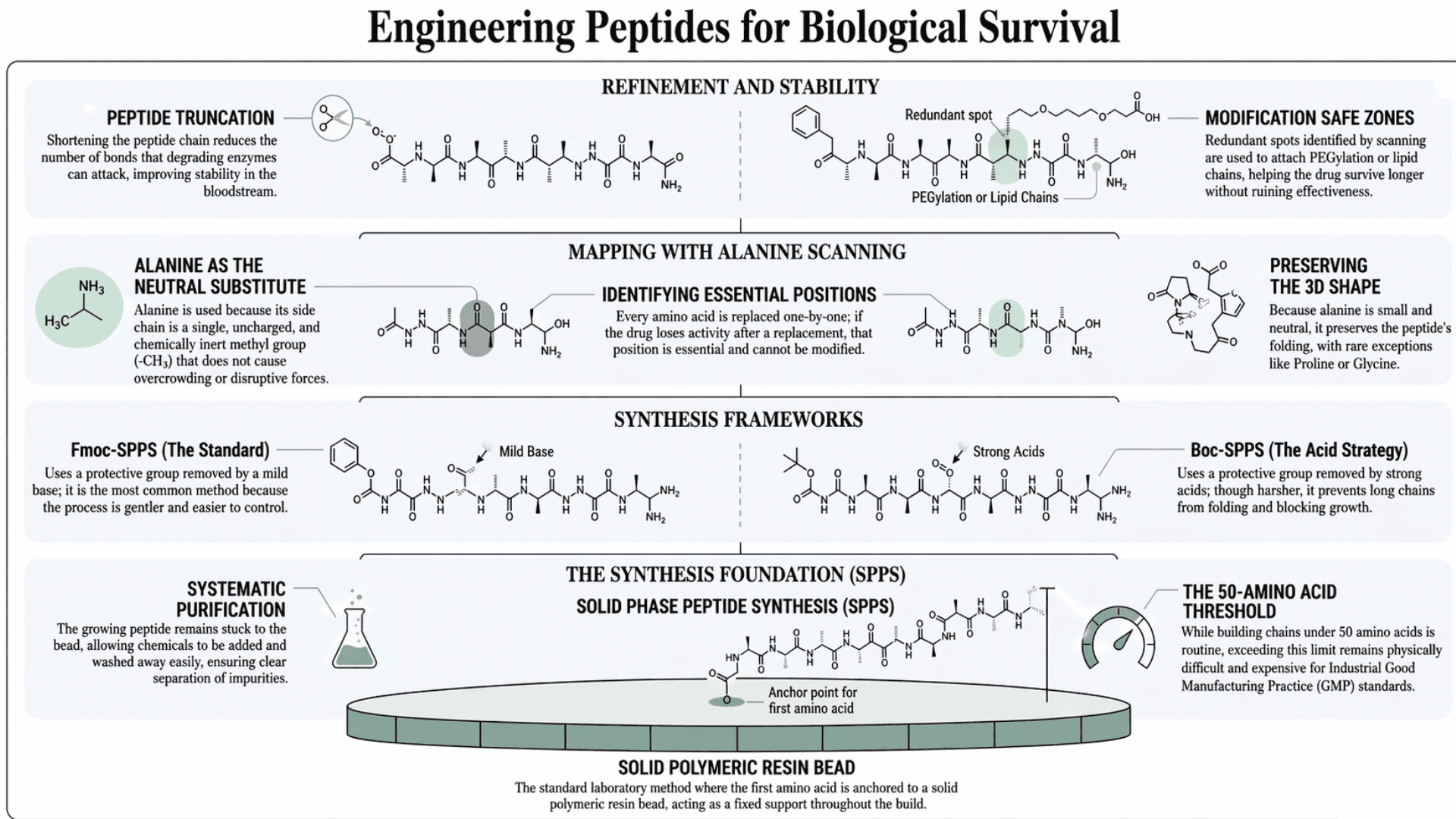
Solid Phase Peptide Synthesis (SPPS) is the standard lab method used to create therapeutic peptides. First developed by Robert Merrifield in 1963, the creation begins by anchoring the first amino acid to a solid polymeric resin (a solid bead made of plastic-like material).
It acts like a fixed support:
the growing peptide stays stuck to it
chemicals can be added and washed away easily
the peptide is only removed at the end
The benefit of this is that it enables a clear separation of the impurities from the developing peptide at each step. This practically makes the purification process simpler than biological methods (Wang et al., Nature).
There are two main strategies:
Fmoc-SPPS (Fluorenylmethyloxycarbonyl Solid-Phase Peptide Synthesis):
Uses a protective group called Fmoc to block part of the amino acid during synthesis. This group is removed using a mild base, making the process gentler and easier to control. It is the most commonly used method today.
Boc-SPPS (tert-Butyloxycarbonyl Solid-Phase Peptide Synthesis):
Utilizes a protective group called Boc, which is removed using strong acids. This method is harsher as it uses acid-based chemistry. But it can be useful for longer peptide chains because it helps prevent the chain from folding in a way that blocks further growth.
Building chains of fewer than 50 amino acids is now relatively routine. Chains exceeding 50 amino acids remain physically difficult and expensive at an industrial scale.
The reason is simple: Good Manufacturing Practice standards require mild reaction conditions to minimize byproducts. And more than that, there is limited access to the equipment needed to use microwave or infrared methods for making peptides on a large commercial scale.
As a result, it makes sense that the more reliable conventional SPPS approach is preferred by most GMP manufacturers. Even if it is slower (Wang et al., Nature).
How do scientists identify which parts of a peptide sequence are essential before modifying it?
Scientists use a technique called alanine scanning before they start changing a peptide. This helps them map which positions in the sequence are essential for biological activity. Alanine is used because it is the perfect neutral substitute. Its side chain is just a single, uncharged, and chemically inert methyl group (-CH3).
Every amino acid in the peptide sequence is replaced one at a time with alanine, a small amino acid with a chemically inert methyl side chain. This does not change the secondary structure. Because alanine is small and neutral, it does not cause steric clash (overcrowding) or introduce new, disruptive electrostatic forces.
Therefore, it preserves the peptide's overall 3D shape and folding. (Note: The only rare exceptions to this rule are Proline, which naturally kinks the peptide backbone, and Glycine, which provides extreme flexibility).
If replacing a specific amino acid causes the drug to lose activity, that position is identified as essential and cannot be modified. If activity is maintained, that position is redundant and available for modification (Fetse et al., NIH).
In drug development, these redundant spots are highly valuable. Scientists use these safe zones to attach chemical modifications (like PEGylation or lipid chains) that help the drug survive longer in the bloodstream without ruining its effectiveness.
Alanine scanning is often followed by peptide truncation. Shortening the chain reduces the number of bonds that degrading enzymes can attack, which improves stability without necessarily losing biological activity. Together, these two techniques allow researchers to design the shortest, most stable version of a sequence that retains full therapeutic function.
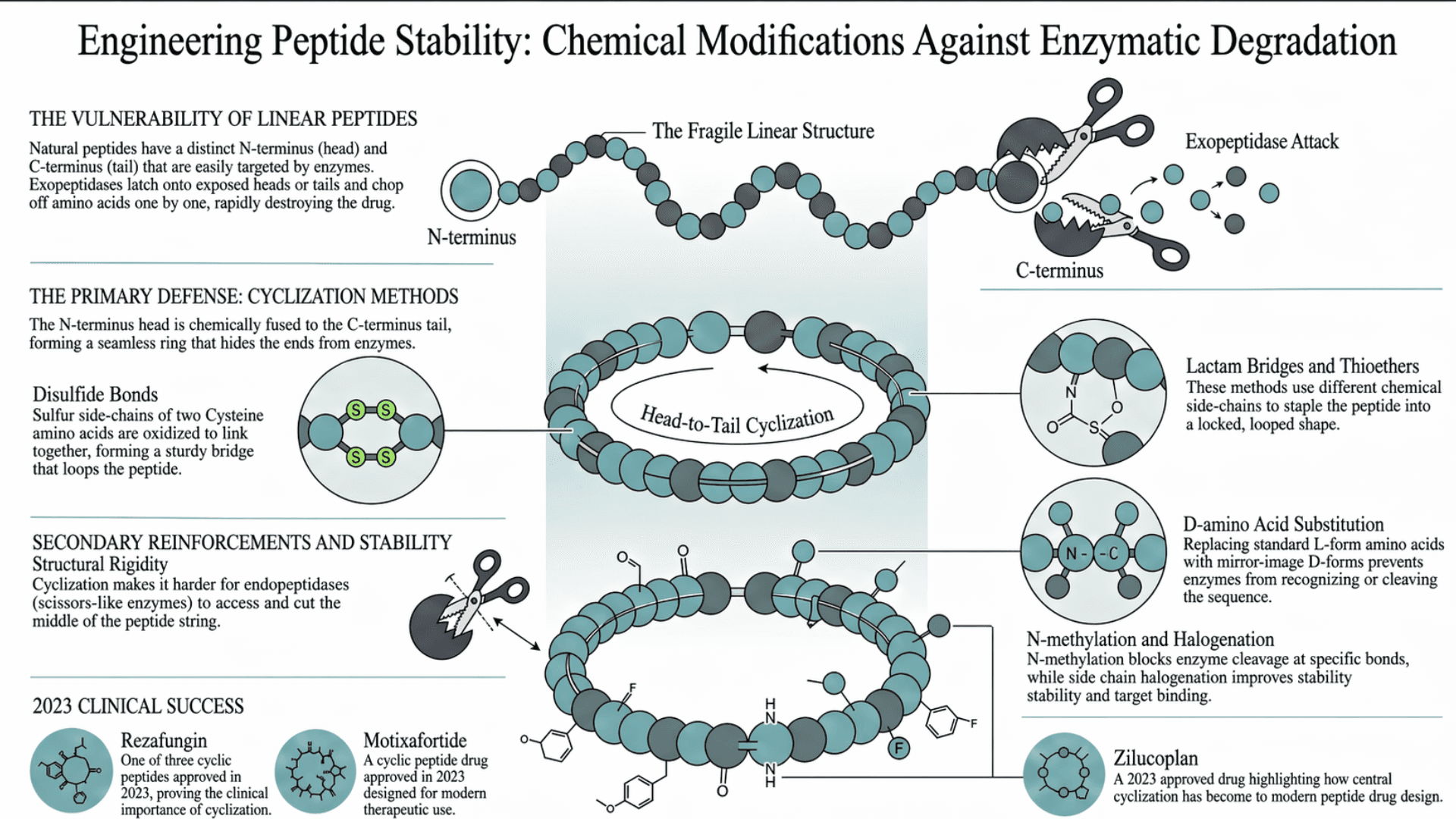
What chemical modifications make a peptide resistant to the enzymes that break it down?
The Problem: Linear Peptides are Fragile
When a peptide is built naturally, it is linear. It has a distinct starting point (the N-terminus, or head) and a distinct ending point (the C-terminus, or tail). When a linear peptide drug is injected into the bloodstream, exopeptidases immediately recognize these exposed ends. They latch onto the head or the tail and begin chopping off amino acids one by one. This rapidly destroys the drug before it can reach its target.
The Solution: Tying the Knot or Cyclization
Cyclization solves this by physically hiding the ends. By chemically bonding the peptide together, the starting points that the enzymes rely on are removed (Zhang et al., ScienceDirect).
Scientists can do this in a number of ways:
Head-to-Tail:
The literal N-terminus head is chemically fused to the C-terminus tail, forming a seamless ring.
Disulfide Bonds:
If the peptide has two Cysteine amino acids, their sulfur side-chains can be oxidized to link together, forming a sturdy bridge that loops a section of the peptide.
Lactam Bridges / Thioethers:
Similar to disulfide bonds, these use different chemical side-chains to staple the peptide into a locked, looped shape.
A Secondary Benefit: Rigidity
Besides exopeptidases (which attack the ends), there is another type of enzyme called an endopeptidase, which acts like scissors. It can cut the middle of the string. Cyclization also makes it harder for enzymes to access and cut the middle of the peptide (Montalbán-López et al., ScienceDirect).
Among the six peptide drugs approved in 2023, three were cyclic peptides: rezafungin, motixafortide, and zilucoplan, underlining how central cyclization has become to modern peptide drug design (Xiao et al., Nature).
Other Chemical Modification Methods:
D-amino acid substitution replaces standard L-form amino acids with their mirror-image D forms at specific positions in the sequence. Digestive enzymes evolved to process L-form amino acids specifically. They cannot efficiently recognize or cleave D-forms at substituted positions, extending the plasma half-life of the drug significantly (Wang et al., Nature), (Iglesias et al., NIH).
N-methylation adds a methyl group to the nitrogen atom of specific peptide bonds. This blocks the enzyme's ability to cleave at those positions. Peptoid formation changes the structure of the peptide backbone so enzymes cannot easily recognize it. Side chain halogenation adds atoms like fluorine or chlorine to improve stability and help the peptide bind better to its target. (Fetse et al., NIH).
What are stapled peptides, and why do they matter for targeting difficult protein interactions?
Proteins often interact using alpha-helix-shaped regions at their contact points. A peptide designed to copy this shape often loses it when it is in solution and unfolds before it reaches its target. This creates a major design problem. It limits how effectively standard peptide drugs can block these protein interactions.
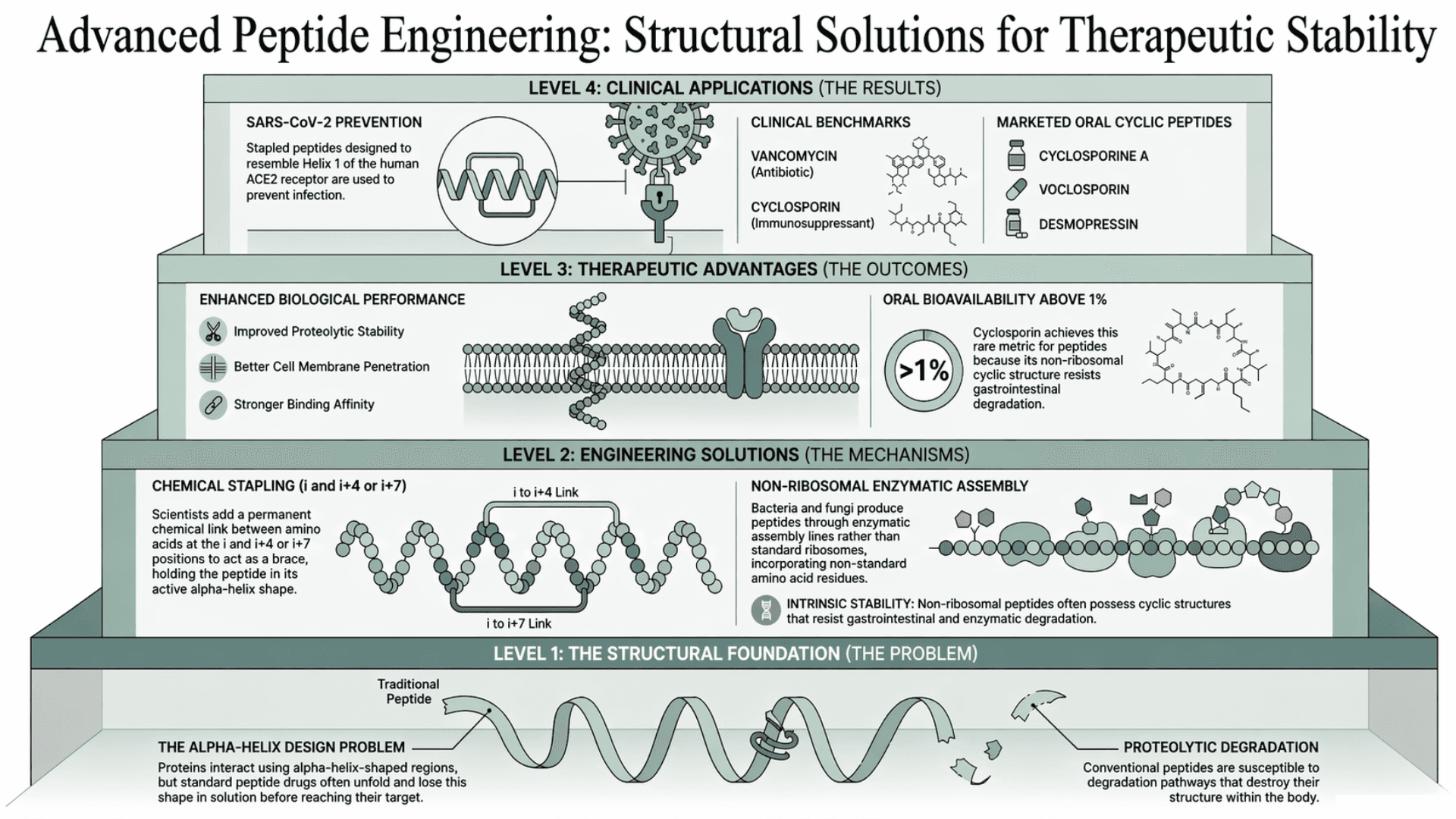
Stapled peptides fix the problem of shape loss by adding a permanent chemical link between two points in the chain, specifically at the i and i+4 or i+7 positions, holding the helix in its active shape.
In a peptide sequence, each amino acid has a position (1, 2, 3, etc.). The terms i and i+4 or i+7 mean:
a. one amino acid at a position (i)
b. another one 4 or 7 steps ahead in the sequence.
Scientists connect these two points with a strong bond. This acts like a brace that holds the peptide in its correct shape (an alpha helix), instead of letting it unfold.
Studies have confirmed that stapled peptides show improved proteolytic stability, better cell membrane penetration, and stronger binding affinity compared to their unmodified counterparts.
An example of clinical application is stapled peptides designed to resemble Helix 1 of the human ACE2 receptor. These have shown varying degrees of efficacy in preventing SARS-CoV-2 infection (Xiao et al., Nature).
What are non-ribosomal peptides, and what makes them structurally distinct?
Non-ribosomal peptides are produced by bacteria and fungi through enzymatic assembly lines rather than through the standard ribosomal process. They contain non-standard amino acid residues that the body's enzymes do not recognize. Thus, they are naturally resistant to the degradation pathways that destroy conventional peptides.
Vancomycin and cyclosporin are the most clinically significant examples. Vancomycin is a glycopeptide antibiotic. Cyclosporin is an immunosuppressant. Both possess cyclic structures with high intrinsic stability. Cyclosporin is one of the few peptides with oral bioavailability above 1%. That’s because its non-ribosomal cyclic structure resists gastrointestinal degradation.
Among the currently marketed oral peptide drugs, three are cyclic: cyclosporine A, voclosporin, and desmopressin (Xiao et al., Nature). Non-ribosomal peptides have many different structures. So they are used as a base to develop drugs for infections and immune diseases. Their features also help scientists design new synthetic peptides.
Therapeutic peptides have evolved from simple biological molecules into precisely engineered drugs. With advances in structure design and chemical modification, many of their original limitations are being overcome. As these technologies improve, peptides are becoming a core part of modern drug development.
FAQ
Are peptide drugs safer than traditional drugs?
Peptide drugs are generally safer because they break down into amino acids that the body can reuse. This reduces the risk of toxic buildup compared to many small molecule drugs.Why are peptide drugs usually injected instead of taken as pills?
Most peptides are quickly broken down in the digestive system and cannot easily pass through the gut lining. Injection allows them to enter the bloodstream before they are destroyed.How long do peptide drugs stay active in the body?
Natural peptides may last only minutes. Modified peptides can remain active for hours or even days, depending on how they are engineered.What makes peptide drugs difficult or expensive to produce?
Peptides are built step by step with high precision. As the chain gets longer, the process becomes more complex, requiring strict quality control and increasing cost.Can peptide drugs target diseases that other drugs cannot?
Yes. Peptides can interact with large protein surfaces that small molecules cannot effectively block, allowing them to target pathways that are otherwise difficult to treat.What are the main challenges in developing peptide drugs?
The biggest challenges are rapid breakdown in the body and difficulty crossing cell membranes. Most design strategies focus on improving stability and delivery.Do peptide drugs trigger immune reactions?
They can in some cases. If the peptide is seen as foreign, the body may produce antibodies against it, which can reduce effectiveness.What is the difference between natural and synthetic peptides?
Natural peptides are produced in the body. Synthetic peptides are engineered to improve stability, activity, and how long they last.Why are peptides considered a middle ground in drug design?
They combine the advantages of both small molecules and biologics. They are large enough for high specificity but small enough to be easier to produce and modify.How widely are peptide drugs used today?
More than 120 peptide and protein-based drugs have been approved globally, and their use continues to expand across multiple disease areas.

